SQLite in Python
SQLite (https://www.sqlite.org\ is a lightweight, SQL/relational database that is available by default with Python (https://docs.python.org/3/library/sqlite3.html\. By using import sqlite3 you can interact with an SQLite database. So, let's create one, returning to our earlier Centrifuge output. Here is the file "tables.sql" containing the SQL statements needed to drop and create the tables:
drop table if exists tax;
create table tax (
tax_id integer primary key,
tax_name text not null,
ncbi_id int not null,
tax_rank text default '',
genome_size int default 0,
unique (ncbi_id)
);
drop table if exists sample;
create table sample (
sample_id integer primary key,
sample_name text not null,
unique (sample_name)
);
drop table if exists sample_to_tax;
create table sample_to_tax (
sample_to_tax_id integer primary key,
sample_id int not null,
tax_id int not null,
num_reads int default 0,
abundance real default 0,
num_unique_reads integer default 0,
unique (sample_id, tax_id),
foreign key (sample_id) references sample (sample_id),
foreign key (tax_id) references tax (tax_id)
);
Like Python, has data types of strings, integers, and floats (https://sqlite.org/datatype3.html\). Primary keys are unique values defining a record in a table. You can place constraints on the allowed values of a field with conditions like default values or not null requirements as well as having the database enforce that some values are unique (such as NCBI taxonomy IDs). You can also require that a particular combination of fields be unique, e.g., the sample/tax table has a unique constraint on the pairing of the sample/tax IDs. Additionally, this database uses foreign keys (https://sqlite.org/foreignkeys.html\) to maintain relationships between tables. We will see in a moment how that prevents us from accidentally creating "orphan" records.
We are going to create a minimal database to track the abundance of species in various samples. The biggest rule of relational databases is to not repeat data. There should be one place to store each entity. For us, we have a "sample" (the Centrifuge ".tsv" file), a "taxonomy" (NCBI tax ID/name), and the relationship of the sample to the taxonomy. I have my own particular naming convention when it comes to relational tables/fields:
- Name tables in the singular, e.g. "sample" not "samples"
- Name the primary key [tablename] + underscore + "id", e.g., "sample_id"
- Name linking tables [table1] + underscore + "to" + underscore + [table2]
- Always have a primary key that is an auto-incremented integer
Here is a simple E/R (entity-relationship) graph of the schema (created with SQL::Translator):
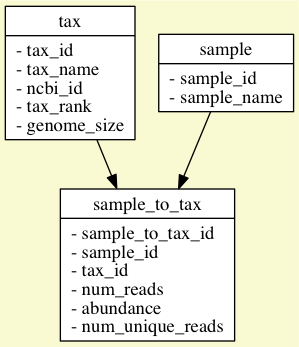
You can instantiate the database by calling make db in the "csv" directory to first remove the existing database and then recreate it by redirecting the "tables.sql" file into sqlite3:
$ make db
find . -name centrifuge.db -exec rm {} \;
sqlite3 centrifuge.db < tables.sql
You can then run sqlite3 centrifuge.db to use the CLI (command-line interface) to the database. Use .help inside SQLite to see all the "dot" commands (they begin with a ., cf. https://sqlite.org/cli.html\):
$ sqlite3 centrifuge.db
SQLite version 3.13.0 2016-05-18 10:57:30
Enter ".help" for usage hints.
sqlite>
I often rely on the .schema command to look at the tables in an SQLite db. If you run that, you should see essentially the same thing as was in the "tables.sql" file. An alternate way to create the database is to use the .read tables.sql command from within SQLite to have it read and execute the SQL statements in that file.
We can manually insert a record into the tax table with an insert statement (https://sqlite.org/lang\_insert.html\). Note how SQLite treats strings and numbers exactly like Python -- strings must be in quotes, numbers should be plain:
sqlite> insert into tax (tax_name, ncbi_id) values ('Homo sapiens', 3606);
We can add a dummy "sample" and link them like so:
sqlite> insert into sample (sample_name) values ('foo');
sqlite> insert into sample_to_tax (sample_id, tax_id, num_reads, abundance)
...> values (1, 1, 100, .01);
Verify that the data is there with a select statement (https://sqlite.org/lang\_select.html\):
sqlite> select count(*) from tax;
1
sqlite> select * from tax;
1|Homo sapiens|3606||0
Use .headers on to see the column names:
sqlite> .headers on
sqlite> select * from tax;
tax_id|tax_name|ncbi_id|tax_rank|genome_size
1|Homo sapiens|3606||0
sqlite> select * from sample;
sample_id|sample_name
1|foo
That's still a bit hard to read, so we can set .mode column to see a bit better:
sqlite> select * from sample;
sample_id sample_name
---------- -----------
1 foo
sqlite> select * from tax;
tax_id tax_name ncbi_id tax_rank genome_size
---------- ------------ ---------- ---------- -----------
1 Homo sapiens 3606 0
sqlite> select * from sample_to_tax;
sample_to_tax_id sample_id tax_id num_reads abundance num_unique_reads
---------------- ---------- ---------- ---------- ---------- ----------------
1 1 1 100 0.01 0
Often what we want is to join the tables so we can see just the data we want, e.g.:
sqlite> select s.sample_name, t.tax_name, s2t.num_reads
...> from sample s, tax t, sample_to_tax s2t
...> where s.sample_id=s2t.sample_id
...> and s2t.tax_id=t.tax_id;
sample_name tax_name num_reads
----------- ------------ ----------
foo Homo sapiens 100
Now let's try to delete the sample record after we have turned on the enforcement of foreign keys:
sqlite> PRAGMA foreign_keys = ON;
sqlite> delete from sample where sample_id=1;
Error: FOREIGN KEY constraint failed
It would be bad to remove our sample and leave the sample/tax records in place. This is what foreign keys do for us. (Other databases -- PostgreSQL, MySQL, Oracle, etc. -- do this without having to explicitly turn on this feature, but keep in mind that this is an extremely lightweight, fast, and easy database to create and administer. When you need more speed/power/safety, then you will move to another database.)
Obviously we're not going to manually enter our data by hand, so let's write a script to import some data. This script is going to be somewhat long, so let's break it down. Here's the start. We need to take as arguments the Centrifuge "*.tsv" (tab-separated values) file which is the summary table for all the species found in a given sample. The script will take one or more of these positional arguments. It will also take as a named argument the --db name of the SQLite database. Note that the sqlite3 module is available by default with Python -- no need to install anything!
#!/usr/bin/env python3
"""Load Centrifuge into SQLite db"""
import argparse
import csv
import os
import re
import sqlite3
import sys
# --------------------------------------------------
def get_args():
"""get args"""
parser = argparse.ArgumentParser(description='Load Centrifuge data')
parser.add_argument('tsv_file', metavar='file',
help='Sample TSV file', nargs='+')
parser.add_argument('-d', '--dbname', help='Centrifuge db name',
metavar='str', type=str, default='centrifuge.db')
return parser.parse_args()
Our main is going to handle the arguments, ensuring the --dbname is a valid file, then processing each of the tsv_file arguments (note the nargs declaration to show that the program takes one or more TSV files). Note that in order to keep this function short, I created two other functions, to import the samples and TSV files:
# --------------------------------------------------
def main():
"""main"""
args = get_args()
tsv_files = args.tsv_file
dbname = args.dbname
if not os.path.isfile(dbname):
print('Bad --dbname "{}"'.format(dbname))
sys.exit(1)
db = sqlite3.connect(dbname)
for fnum, tsv_file in enumerate(tsv_files):
if not os.path.isfile(tsv_file):
print('Bad tsv_file "{}"'.format(tsv_file))
sys.exit(1)
sample_name, ext = os.path.splitext(tsv_file)
if ext != '.tsv':
print('"{}" does not end with ".tsv"'.format(tsv_file))
sys.exit(1)
if sample_name.endswith('.centrifuge'):
sample_name = re.sub(r'\.centrifuge$', '', sample_name)
sample_id = import_sample(sample_name, db)
print('{:3}: Importing "{}" ({})'.format(fnum + 1,
sample_name, sample_id))
import_tsv(db, tsv_file, sample_id)
print('Done')
Here is the code to import a "sample." It needs a sample_name (which we assume to be unique) and a database handle (which is a bit like filehandles which we've been dealing with -- it's the actual conduit from your code to the database). First we have to check if the sample already exists in our table, and this requires we use a cursor (https://docs.python.org/3/library/sqlite3.html\) to issue our select statement. Rather than putting the sample name directly into the SQL (which is very insecure, see SQL injection/"Bobby Tables" XKCD https://xkcd.com/327\), we use a ? and pass the string as an argument to the execute function. If nothing (None) is returned, we can safely insert the new record and get the newly created sample ID from the lastrowid function of the cursor; otherwise, the sample ID is in the res result list as the first field:
# --------------------------------------------------
def import_sample(sample_name, db):
"""Import sample"""
cur = db.cursor()
cur.execute('select sample_id from sample where sample_name=?',
(sample_name,))
res = cur.fetchone()
if res is None:
cur.execute('insert into sample (sample_name) values (?)',
(sample_name,))
sample_id = cur.lastrowid
else:
sample_id = res[0]
return sample_id
The code to import the TSV file is similar. We establish SQL statements to find/insert/update the sample/tax record, then we use the csv module to parse the TSV file, creating dictionaries of each record (a product of merging the first line/headers with each row of data). Again, to keep this function short enough to fit on a "page," there is a separate function to find or create the taxonomy record.
# --------------------------------------------------
def import_tsv(db, file, sample_id):
"""Import TSV file"""
find_sql = """
select sample_to_tax_id
from sample_to_tax
where sample_id=?
and tax_id=?
"""
insert_sql = """
insert
into sample_to_tax
(sample_id, tax_id, num_reads, abundance, num_unique_reads)
values (?, ?, ?, ?, ?)
"""
update_sql = """
update sample_to_tax
set sample_id=?, tax_id=?, num_reads=?,
abundance=?, num_unique_reads=?
where sample_to_tax_id=?
"""
cur = db.cursor()
with open(file) as csvfile:
reader = csv.DictReader(csvfile, delimiter='\t')
for row in reader:
tax_id = find_or_create_tax(db, row)
if tax_id:
cur.execute(find_sql, (sample_id, tax_id))
res = cur.fetchone()
num_reads = row.get('numReads', 0)
abundance = row.get('abundance', 0)
num_uniq = row.get('numUniqueReads', 0)
if res is None:
cur.execute(insert_sql,
(sample_id, tax_id, num_reads,
abundance, num_uniq))
else:
s2t_id = res[0]
cur.execute(update_sql,
(sample_id, tax_id, num_reads,
abundance, num_uniq, s2t_id))
else:
print('No tax id!')
db.commit()
return 1
The find/create tax function works just the same as that for the sample:
# --------------------------------------------------
def find_or_create_tax(db, rec):
"""find or create the tax"""
find_sql = 'select tax_id from tax where ncbi_id=?'
insert_sql = """
insert into tax (tax_name, ncbi_id, tax_rank, genome_size)
values (?, ?, ?, ?)
"""
cur = db.cursor()
ncbi_id = rec.get('taxID', '')
if re.match('^\d+$', ncbi_id):
cur.execute(find_sql, (ncbi_id,))
res = cur.fetchone()
if res is None:
name = rec.get('name', '')
if name:
print('Loading "{}" ({})'.format(name, ncbi_id))
cur.execute(insert_sql,
(name, ncbi_id, rec['taxRank'],
rec['genomeSize']))
tax_id = cur.lastrowid
else:
print('No "name" in {}'.format(rec))
return None
else:
tax_id = res[0]
return tax_id
else:
print('"{}" does not look like an NCBI tax id'.format(ncbi_id))
return None
If you use make data, several files will be downloaded from the iMicrobe FTP site for use by the make load step run the loader program:
$ make load
./load_centrifuge.py *.tsv
1: Importing "YELLOWSTONE_SMPL_20717" (1)
Loading "Synechococcus sp. JA-3-3Ab" (321327)
Loading "Synechococcus sp. JA-2-3B'a(2-13)" (321332)
2: Importing "YELLOWSTONE_SMPL_20719" (2)
Loading "Streptococcus suis" (1307)
Loading "synthetic construct" (32630)
3: Importing "YELLOWSTONE_SMPL_20721" (3)
Loading "Staphylococcus sp. AntiMn-1" (1715860)
4: Importing "YELLOWSTONE_SMPL_20723" (4)
5: Importing "YELLOWSTONE_SMPL_20725" (5)
6: Importing "YELLOWSTONE_SMPL_20727" (6)
Done
Now we can inspect how many records were loaded into the database:
$ sqlite3 centrifuge.db
SQLite version 3.13.0 2016-05-18 10:57:30
Enter ".help" for usage hints.
sqlite> select count(*) from tax;
5
sqlite> select count(*) from sample;
6
sqlite> select count(*) from sample_to_tax;
18
But, again, we're not going to just sit here and manually write SQL to check out the data. Let's write a program that takes an NCBI tax id as an argument and reports the samples where it is found. You will need to make tabulate to run the command to install the "tabulate" module (https://pypi.python.org/pypi/tabulate\) in order to run this program:
1 #!/usr/bin/env python3
2 """Query centrifuge.db for NCBI tax id"""
3
4 import argparse
5 import os
6 import re
7 import sys
8 import sqlite3
9 from tabulate import tabulate
10
11 # --------------------------------------------------
12 def get_args():
13 """get args"""
14 parser = argparse.ArgumentParser(description='Argparse Python script')
15 parser.add_argument('-d', '--dbname', help='Centrifuge db name',
16 metavar='str', type=str, default='centrifuge.db')
17 parser.add_argument('-o', '--orderby', help='Order by',
18 metavar='str', type=str, default='abundance')
19 parser.add_argument('-s', '--sortorder', help='Sort order',
20 metavar='str', type=str, default='desc')
21 parser.add_argument('-t', '--taxid', help='NCBI taxonomy id',
22 metavar='str', type=str, required=True)
23 return parser.parse_args()
24
25 # --------------------------------------------------
26 def main():
27 """main"""
28 args = get_args()
29 dbname = args.dbname
30 order_by = args.orderby
31 sort_order = args.sortorder
32
33 if not os.path.isfile(dbname):
34 print('"{}" is not a valid file'.format(dbname))
35 sys.exit(1)
36
37 flds = set(['tax_name', 'num_reads', 'abundance', 'sample_name'])
38 if not order_by in flds:
39 print('"{}" not an allowed --orderby, choose from {}'.format(
40 order_by, ', '.join(flds)))
41 sys.exit(1)
42
43 sorting = set(['asc', 'desc'])
44 if not sort_order in sorting:
45 print('"{}" not an allowed --sortorder, choose from {}'.format(
46 order_by, ', '.join(sorting)))
47 sys.exit(1)
48
49 tax_ids = []
50 for tax_id in re.split(r'\s*,\s*', args.taxid):
51 if re.match(r'^\d+$', tax_id):
52 tax_ids.append(tax_id)
53 else:
54 print('"{}" does not look like an NCBI tax id'.format(tax_id))
55
56 if len(tax_ids) == 0:
57 print('No tax ids')
58 sys.exit(1)
59
60 db = sqlite3.connect(dbname)
61 cur = db.cursor()
62 sql = """
63 select s.sample_name, t.tax_name, s2t.num_reads, s2t.abundance
64 from sample s, tax t, sample_to_tax s2t
65 where s.sample_id=s2t.sample_id
66 and s2t.tax_id=t.tax_id
67 and t.ncbi_id in ({})
68 order by {} {}
69 """.format(', '.join(tax_ids), order_by, sort_order)
70
71 cur.execute(sql)
72
73 samples = cur.fetchall()
74 if len(samples) > 0:
75 cols = [d[0] for d in cur.description]
76 print(tabulate(samples, headers=cols))
77 else:
78 print('No results')
79
80 # --------------------------------------------------
81 if __name__ == '__main__':
82 main()
It takes as arguments a required NCBI tax id that can be a single value or a comma-separated list. Options include the SQLite Centrifuge db, a column name to sort by, and whether to show in ascending or descending order. The output is formatted with the tabulate module to produce a simple text table. To query by one tax ID:
$ ./query_centrifuge.py -t 321327
sample_name tax_name num_reads abundance
---------------------- -------------------------- ----------- -----------
YELLOWSTONE_SMPL_20721 Synechococcus sp. JA-3-3Ab 315 0.98
YELLOWSTONE_SMPL_20723 Synechococcus sp. JA-3-3Ab 6432 0.98
YELLOWSTONE_SMPL_20727 Synechococcus sp. JA-3-3Ab 1219 0.96
YELLOWSTONE_SMPL_20717 Synechococcus sp. JA-3-3Ab 19 0.53
YELLOWSTONE_SMPL_20719 Synechococcus sp. JA-3-3Ab 719 0.27
YELLOWSTONE_SMPL_20725 Synechococcus sp. JA-3-3Ab 3781 0.2
To query by more than one:
$ ./query_centrifuge.py -t 321327,1307
sample_name tax_name num_reads abundance
---------------------- -------------------------- ----------- -----------
YELLOWSTONE_SMPL_20721 Synechococcus sp. JA-3-3Ab 315 0.98
YELLOWSTONE_SMPL_20723 Synechococcus sp. JA-3-3Ab 6432 0.98
YELLOWSTONE_SMPL_20727 Synechococcus sp. JA-3-3Ab 1219 0.96
YELLOWSTONE_SMPL_20717 Synechococcus sp. JA-3-3Ab 19 0.53
YELLOWSTONE_SMPL_20719 Synechococcus sp. JA-3-3Ab 719 0.27
YELLOWSTONE_SMPL_20725 Synechococcus sp. JA-3-3Ab 3781 0.2
YELLOWSTONE_SMPL_20719 Streptococcus suis 1 0
To order by "num_reads" instead of "abundance":
$ ./query_centrifuge.py -t 321327,1307 -o num_reads
sample_name tax_name num_reads abundance
---------------------- -------------------------- ----------- -----------
YELLOWSTONE_SMPL_20723 Synechococcus sp. JA-3-3Ab 6432 0.98
YELLOWSTONE_SMPL_20725 Synechococcus sp. JA-3-3Ab 3781 0.2
YELLOWSTONE_SMPL_20727 Synechococcus sp. JA-3-3Ab 1219 0.96
YELLOWSTONE_SMPL_20719 Synechococcus sp. JA-3-3Ab 719 0.27
YELLOWSTONE_SMPL_20721 Synechococcus sp. JA-3-3Ab 315 0.98
YELLOWSTONE_SMPL_20717 Synechococcus sp. JA-3-3Ab 19 0.53
YELLOWSTONE_SMPL_20719 Streptococcus suis 1 0
To sort ascending:
$ ./query_centrifuge.py -t 321327,1307 -o num_reads -s asc
sample_name tax_name num_reads abundance
---------------------- -------------------------- ----------- -----------
YELLOWSTONE_SMPL_20719 Streptococcus suis 1 0
YELLOWSTONE_SMPL_20717 Synechococcus sp. JA-3-3Ab 19 0.53
YELLOWSTONE_SMPL_20721 Synechococcus sp. JA-3-3Ab 315 0.98
YELLOWSTONE_SMPL_20719 Synechococcus sp. JA-3-3Ab 719 0.27
YELLOWSTONE_SMPL_20727 Synechococcus sp. JA-3-3Ab 1219 0.96
YELLOWSTONE_SMPL_20725 Synechococcus sp. JA-3-3Ab 3781 0.2
YELLOWSTONE_SMPL_20723 Synechococcus sp. JA-3-3Ab 6432 0.98